
Introdução
“Nós não sabemos exatamente o que causa o tipo de fenilcetonúria da qual Charlie sofria desde criança: algum tipo de ocorrência bioquímica ou genética, possivelmente radiação ionizante, radiação natural ou até um ataque de vírus ao feto; quer que seja resultou em um gene defeituoso que produz uma, digamos assim, “enzima dissidente” que cria reações bioquímicas defeituosas. E é claro, aminoácidos recentemente produzidos competem com enzimas normais, causando dano neurológico.”
Esse é um trecho retirado do livro “Flores para Algernon” de Daniel Keyes, escrito em 1959. A história centra-se em Charlie Gordo, deficiente mental por uma fenilcetonúria não tratada. No livro, Charlie é um cobaia humano para a cirurgia que tem a intenção aumentar suas habilidades intelectuais. O presente artigo tem o intuito de abordar as alterações metabólicas decorrente da fenilcetonúria que impactam neurofisiologicamente, explicando assim a sua associação com a deficiência mental.
Vias metabólicas
A conversão de Phe em Tyr ocorre por um sistema de hidroxilação que depende de inúmeros fatores que serão detalhados a seguir.
Analisando a estrutura química do α-aminoácido Phe sabemos que ele tem uma cadeia lateral apolar e por ser eletricamente neutro facilita interações hidrofóbicas. Ele é percursor da Tirosina, que também é α-aminoácido com um caráter polar devido a cadeia lateral cíclica aromática com um grupo álcool.
Em 2001 Scriver CR e Kaufman S relataram a existência de enantiômeros do Phe, sendo que a ingestão de L-Phe (levogiro) ocorre por meio da dieta de aminoácidos. A hidroxilação por PAH com seu cofator BH4 (tetrahidrobiopterina) na presença de oxigênio, produz L-Tyr (levogiro). O metabolismo alternativo de L-Phe por descarboxilação ou transaminação produz vários metabólitos que são excretados na urina.
Para hidroxilação do Phe é necessário que ocorra a lise do anel benzênico. A via de transaminação e descarboxilação forma metabólitos, como fenilpiruvato, fenillactato, e o- hidroxifenilacetato que são posteriormente excretados pela via urinária.
Distúrbios que afetam o sistema de regeneração da BH4 (tetrahidrobiopterina), como alterações na dihidropteridina redutase e 4α-carbinolamina desidratase afetam homeostaticamente a biossíntese de catecolaminas e serotonina (O triptofano é um aminoácido importante na síntese de serotonina).
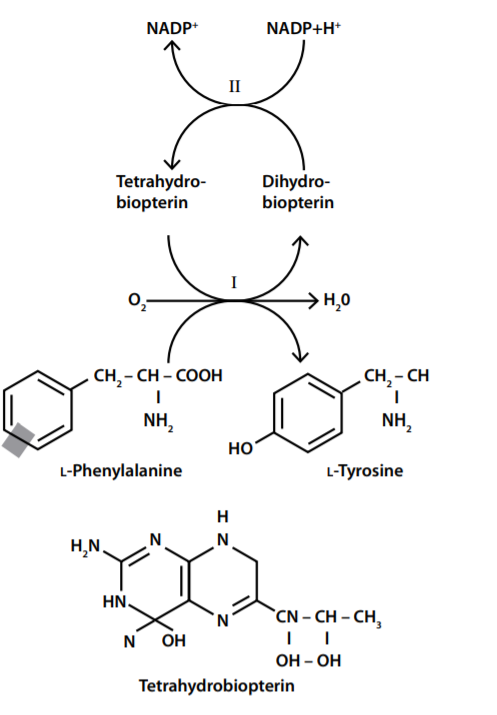
Com base no que foi supracitado, sabemos que a conversão da fenilalanina para tirosina depende da oxidação da tetrahidrobiopterina para dihidropteridina quinóide e sua redução em dihidropteridina redutase. Com essa premissa, podemos explicar uma das causas de hiperfenilalaninemia por deficiência de tetrahidrobiopterina, uma doença genética autossómica recessiva, conhecida como deficiência de diidropteridina redutase. Essa condição também afeta a neurotransmissão de monoaminas, pois alterações na tetrahidrobiopterina levam a uma disfunção na triptofano hidroxilase. A diminuição dos níveis da seratonina pode estar relacionada ao efeito da alta concentração de Phe no transporte de aminoácidos através da barreira hematoencefálica ou em enzimas envolvidas na síntese de neurotransmissores. Phe também é um inibidor competitivo da hidroxilase tirosina e da hidroxilase triptofano enzimas importantes para a síntese cerebral dos neurotransmissores dopamina e serotonina, respectivamente. Esses efeitos colaboram para o comprometimento das catecolaminas no cérebro encontradas em pacientes com Fenilcetonúria.
A hidroxilação estereoespecífica da Phe é catalisada pela Fenilalanina Hidroxilase (PAH) que tem a função de transferir um átomo de oxigênio para o anel aromático da fenilalanina. Ela pertence a classe das monooxigenases que dependem da pterina e utilizam tetrahidrobiopterina 4 como cofator e um um ferro não heme para catálise. A Fenilalanina Hidroxilase existe como uma mistura de tetrâmeros e dímeros. No seu domínio regulador, existe um resíduo de serina que se pensa estar envolvido na ativação por fosforilação.
Genética da Fenilcetonúria
Estudos indicam que pacientes com Fenilcetonúria ao serem submetidos a ressonância magnética podem apresentar alterações na substância branca no cérebro que estão relacionadas a concentração de Phe no seu sangue. Apesar do quociente de inteligência (QI) depender de diversos parâmetros, estudos relataram que existe uma associação entre a gravidade das mutações no gene PAH e esse quociente de inteligência, nesse parágrafo, vamos detalhar o mecanismo responsável por esse fenômeno.
O gene humano da Fenilalanina Hidroxilase está localizado no cromossomo 12q23.2 e mutações no locus PAH (esse gene é bialélico apresenta grande variação alélica e na patogenia da Fenilcetonúria foram encontradas alterações em todos os seus 13 exons) ou mutações nos loci que afetam a síntese e regeneração tetrahidrobiopterina 4 (a hidroxilação por PAH também depende da disponibilidade de seu cofator BH4) em são responsáveis pelo aparecimento da Fenilcetonúria. Essas mutações patogênicas ocorrem alteram a estrutura e função da enzima. O BH4 é biossintetizado de novo a partir de sua molécula precursora de trifosfato de guanosina através de reações químicas dependentes de fosfato de dinucleotídeo de nicotinamida, Zn+2 e Mg+2.
O BH4 é um catalisador para a óxido nítrico sintase durante a síntese da molécula mensageira óxido nítrico a partir do aminoácido arginina. Outra função do BH4 é catalisar a hidroxilação do alquilglicerol pela alquilglicerol monooxigenase. Portanto, a biossíntese insuficiente deste cofator crítico pode levar ao desenvolvimento da Fenilcetonúria por uma disfunção neurovascular.
Estudos também sugerem que alterações na homeostase do cálcio também estejam relacionadas com a patogênse da Fenilcetonúria: estudos sugerem que ocorre um aumento do hormônio paratireóideo e do desidrocolecalciferol em pacientes com essa patologia. Além disso, o O Phe altera as concentrações intracelulares de cálcio livre modulando a Ca+2 -ATPase da membrana plasmática em neurônios corticais.
Impacto neurofisiológico e a Deficiência Intelectual
Como foi abordado acima, o efeito das mutações da Fenilalanina Hidroxilase está relacionado com alterações no desenvolvimento do sistema nervoso central e a concentrações de Phe estão relacionadas com a neuropatologia por um processo de desmielinização.
A deficiência de Tirosina e seus componentes metabólitos através da barreia hematoencefálica tem alterações na neuroquímica e o metabolismo dos percurssores da dopamina e suas catecolaminas. Na presença de concentrações elevadas de Phe, pode ocorrer um bloqueio no transporte da tirosina e do triptofano, afetando a seratonina e as catecolaminas.
O texto é de total responsabilidade do autor e não representa a visão da sanar sobre o assunto.
Observação: material produzido durante vigência do Programa de colunistas Sanar junto com estudantes de medicina e ligas acadêmicas de todo Brasil. A iniciativa foi descontinuada em junho de 2022, mas a Sanar decidiu preservar todo o histórico e trabalho realizado por reconhecer o esforço empenhado pelos participantes e o valor do conteúdo produzido. Eventualmente, esses materiais podem passar por atualização.
Novidade: temos colunas sendo produzidas por Experts da Sanar, médicos conceituados em suas áreas de atuação e coordenadores da Sanar Pós.
Referências
National Institute of Health Consensus Development Conference Statement – Phenylketonuria: Screening and Management. Pediatrics 2001. 108:972-982
Eisensmith RC, Woo SLC. Phenylketonuria. In: CONNEALLY, P. M. (ed). Molecular basis of neurology. Blackwell Scientific Publications, 1983,181-198
Amorim T, Gatto SPP, Boa-Sorte N, Leite MEQ, Fontes MIMM, Barretto J, et al. Aspectos clínicos da fenilcetonúria em serviço de referência em triagem neonatal da Bahia. Rev. Bras. Saúde Matern. Infant, Recife, 5 (4): 457- 462, out. / dez., 2005
Battistini S, Stefano N, Parlanti S, Federico A. Unexpected white matter changes in an early treated PKU case and improvement after dietary treatment. Functional Neurol. 1991; 6(2):177-80.
Sarkissian CN, Gámez, A. Phenylalanine ammonia lyase, enzyme substitution therapy for phenylketonuria, where are we now? Mol Gen Metab. 2005, 86(Suppl 1):22-6.
Giugliani R, Costa JC, Dutra-Filho CS, Niederwieser A. Tetrahydrobiopterin deficiency in two patients with phenylketonuria who did not respond adequately to dietary treatment. Rev Bras Genet 1983;6:557-64.








